
What Do We (Really) Know About DNA Quality For In Vitro Transcription?
By Philip Probert, CPI

The manufacturing of gene therapies and nucleic acid medicines are dependent upon a starting DNA template that defines the protein or antigen that will have the medicinal effect in vivo. Understandably, the sequence of this template DNA being correct is critical to the safety and efficacy of the generated product and is accordingly strictly controlled and measured. Beyond this, other quality attributes of template DNA are recognized to be sources of variability in product quality due to how they impact performance of the template in the manufacturing process(es). For mRNA, what are these attributes, and do we have an absolute understanding of how this template DNA quality impacts the quality of generated mRNA?
Template Specifications To Date
Much of what we understand about DNA template quality is a result of data gained through the application of template DNA for viral vector generation, which is used in transient transfection of cells. For these, a range of different specifications have been proposed to enable the consistent generation of high-quality products though formal requirements have not been defined by either the FDA or EMA to date. For DNA as a starting material for mRNA generation, proposed quality specifications broadly mirror these, similarly with no strict guidance from regulators to date on specifications.1
BioPhorum (2020)2 summarizes a common list of release criteria for GMP-grade plasmid DNA for gene and cell therapy applications. Typically, key indicators of DNA quality are identity, measured through migration by electrophoresis and sequencing, and purity, measured through various impurity assays and confirmation of DNA homogeneity through electrophoresis methods. Cell-derived plasmid DNA, historically the only source of template DNA used as a starting material, is purified as a selection of isoforms, including supercoiled, open circular, and linear. For plasmid DNA, typically, high-quality DNA is considered to have a high percentage (>80%) of supercoiled DNA versus other isoforms. This is on the basis that in order to be supercoiled, plasmid DNA cannot be nicked, linearized, or single-stranded. For synthetic DNA, purity measurements are typically restricted to measurement of migrating species, typically by agarose gel electrophoresis or capillary electrophoresis (CE), given that they do not supercoil.
Consistent Inconsistencies
As an organization, CPI works with a breadth of different modalities and therefore handles DNA from both cell-based and enzymatic-based processes. We routinely work with different DNA sources and types (plasmid and synthetic). With mRNA (and other enzyme-based processes) what we have seen is inconsistent performance of DNA that achieves a “standard” specification but does not deliver a consistent quality of mRNA product. In some instances, this is apparent when measuring mRNA integrity through CE or reverse-phase high-pressure liquid chromatography (RP-HPLC) or when high-grade mRNA is unable to be expressed in cells. Two examples are illustrated here to give further context:
- The percentage of plasmid DNA that is supercoiled is expected to correlate to mRNA quality based on a high supercoiled DNA percentage being part of plasmid DNA quality specifications. Based on this, in the DNA shown in Figure 1, it would be expected that DNA constructs 5 and 6, which had the highest supercoiled percentage, would give rise to higher-quality mRNA than other constructs. However, no apparent difference was noted and, indeed, other examples of customer-based projects have seen “low-quality” DNA giving rise to mRNA with good or better integrity. This is counter to that seen by Piao et al.,3 in which a direct relationship between supercoiled DNA and mRNA quality was seen.
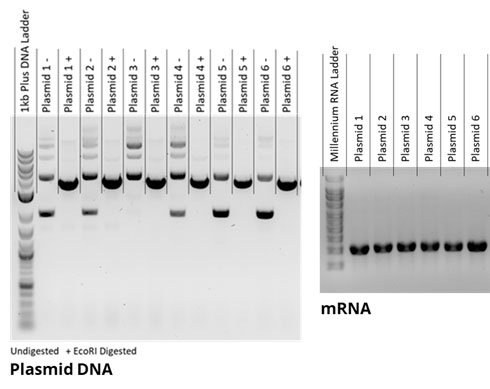
Figure 1: Agarose gel electrophoresis of six different plasmid DNA constructs pre- (-) and post-linearization with EcoRI (+), with mRNA generated through in-vitro transcription shown on right.
- Plasmid DNA was received from a vendor, delivered according to the “standard” DNA quality criteria. This was taken through to in vitro transcription (IVT) to generate mRNA alongside material generated through a DNA gigaprep (internal). As shown in Figure 2, by agarose gel electrophoresis, the DNA looked equivalent in terms of the “standard” specification. The yield, however, was markedly lower in the vendor-supplied plasmid DNA versus that generated internally (approximately 30% lower).
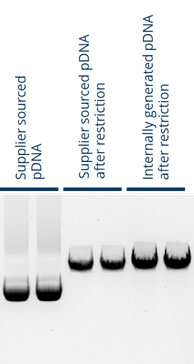
Figure 2: Agarose gel electrophoresis of the same template plasmid DNA (pDNA) received from an external supplier and that generated internally through a gigaprep. Two samples were restricted in preparation of use in IVT.
Why Are We Seeing Inconsistencies?
Much of what we know about DNA quality is based on cell-based systems. A typical use case is transient transfection for viral vector generation. Our experience suggests that DNA suitable for viral vector generation may not always give consistent performance in an IVT. An interpretation of this is that cell-based and cell-free systems are not entirely analogous. In cells, if low-quality DNA is introduced into a cell, it may be that endogenous mechanisms either repair or degrade the DNA or that it is giving rise to low-quality product, but standard analytical methods are insufficient to detect this. In an IVT, the polymerase has no ability to adapt to low-quality template DNA, meaning the quality of the mRNA is directly related to the quality of the DNA. which may not be the case in cell systems. What is true for cell-based applications may therefore not be sufficient or suitable for cell-free applications. This is particularly true given that template DNA for IVT reactions must be linearized prior to use, which is not universally true of use of DNA in cell-based applications.
More generally, in terms of why DNA may be inconsistent, within processes for the generation of plasmid DNA or synthetic DNA there is significant variability, not only within the upstream component of the process where the crude DNA is amplified through cells or enzymatically, but also through to the purification process with different clarification, chromatography, precipitation, and filtration steps. In plasmid DNA purification, variability arising from the alkaline lysis step is known to impact the degree of supercoiling of generated DNA.4 For all DNA, the purification process may involve significant shear, which may damage DNA and is likely a under-recognized source of variability. Different purification approaches also introduce different process related impurities that can also impact IVT performance if not sufficiently cleared.
The final factor that may drive such variability is the DNA sequence itself. It is expected that some sequence elements will impact an IVT reaction. This is undoubtedly a source of variability. Having seen the same sequence manufactured through different processes have variable performance (Figure 2), the extent to which specific sequences are introducing variability is unclear given the variability from the process used to generate them. Greater understanding of the biophysical properties of template DNA used for IVT reactions and contribution to reaction variability should enable sequence-specific effects to be more clearly understood and problematic sequences designed out.
What Are We Not Measuring?
Despite the long list of quality criteria typically used for DNA as a starting material, there remains a reliance on agarose gel electrophoresis or CE for assessment of DNA integrity. Though these enable fragmentation or degradation to be measured, resolution is hugely variable, particularly for agarose gel electrophoresis. As a subject expert reinforced to me recently, a gel can be made to show anything. Furthermore, DNA nicking or low levels of shear resulting are not detectable through these methods. Supercoiling is used as a surrogate marker of DNA integrity, and it enables some conclusions on plasmid DNA quality to be drawn. Unfortunately, this isn’t an approach that can be applied to linearized material or any synthetic DNA, which are the starting points of all IVT reactions – this is a key gap in our analytical toolbox. We are therefore able to make some determination of plasmid DNA quality pre-linearization and can measure fragmentation and/or degradation that is within the resolving power of the electrophoresis methods used. We cannot measure DNA nicking in any linearized DNA or low levels or degradation. These latter attributes may well account for the variability seen in mRNA yield and quality, with damage and degradation giving rise to truncated mRNA species alongside potential impacts to yield. Interestingly, considering the impact of DNA nicking, there is work showing that nicked DNA may be a superior template for PCR than linear DNA.5 If such an effect is true of T7 DNA polymerase, then nicked DNA templates may give rise to a disproportionate quantity of mRNA versus intact DNA.
To address unmeasured impurities being a source of variability, it doesn’t appear likely that these are causing the inconsistencies being seen. Our experience indicates that where template DNA is giving poor yields or quality of mRNA, further washing of the DNA has not improved performance, which suggests the gap is in the quality of the DNA itself.
If fragmentation and DNA damage or nicking are likely to be the main sources of variability, how can we improve our measurement techniques to understand the warts and all of our DNA quality? First, for measurement of fragmentation and degradation, moving from agarose gel electrophoresis to CE is a must. Not all CE systems deliver the same level of resolution, with there often being a trade-off between throughput and resolution. In our labs, the system we used for high-throughput CE doesn’t deliver the same level of resolution as our lower-throughput high-resolution system. An alternative to CE is HPLC-based analysis, such as, for example, anion exchange analysis, which we find to be more informative with regard to DNA quality.
Assessment of DNA damage/nicking is difficult, owing to it being undetectable through separation-based approaches. However, next-generation sequencing methods have been shown to be able to detect such nicks in DNA in research applications.6 This would be a suitable approach given the application of sequencing for determination of template DNA identity and, alongside nick detection, it could also be applied to measure degradation based on sequence coverage. With this, the application of such sequencing pre- and post-linearization would also allow for the quality of the input DNA, rather than the pre-linearized material, to be fully understood ahead of use, which should reduce variability in IVT reactions. This should be further enabled by the rapid continued development of sequencing methods and associated bioinformatics. The more routine quantification of nicking and use of higher-resolution separation-based techniques on DNA post-linearization would allow for DNA from different sources to be more readily substituted and improve understanding of its major sources.
Importantly, with a more complete understanding of DNA quality being utilized in IVT reactions, the degree of analytical characterization required once the process is defined should reduce to just those critical attributes required to ensure generation of mRNA at the required quality.
Future Progress
The importance of template DNA for medicine manufacture continues to increase, alongside the need for it to be of high quality and available on short time frames. DNA quality specifications, however, have not markedly changed over recent years despite the recognition of the criticality of DNA as a starting material, given its impact on product quality. DNA quality measurements are in many cases still reliant upon low-resolution assays and with lack of agreed cohesive specification to define what high-quality DNA is. With the growth of DNA as a starting material and an API in its own right, it is expected that these specifications will become more strictly defined over time and assays improved or introduced. With linearization and purification being sources of further variability, and with linearized template DNA being a common form used as starting material for manufacturing, additional consideration should also be given to other measures for this form of template DNA. This process will be accelerated by more open dialogue about the challenges seen through variability arising from DNA and, with greater data, the impact of sequence on mRNA quality should become clearer. Related to management of variability, use of synthetic DNA over cell-derived plasmid DNA should also give greater consistency of source material, through removal of the biological variability inherent in cell systems.
Until such improvements to methods and specifications are realized, the only reliable method to confirm best performing DNA for a given mRNA manufacturing process is through empirical comparison, whether that be plasmid DNA or synthetic. This will enable the quality of template DNA arising from a particular manufacturing approach to be determined. When this is identified, it will provide a defined starting point for all mRNA generation. If there is any change in the process used in the generation of the template DNA, or its supplier, then performance should be confirmed through mRNA generation and potency assay.
References
- https://www.bioprocessonline.com/doc/what-proposed-plasmid-dna-standards-say-for-gmp-non-gmp-production-0001
- https://www.biophorum.com/download/raw-materials-cell-and-gene-therapy-critical-starting-material-a-discussion-to-help-establish-release-specifications-for-plasmids-and-the-bacterial-master-cell-banks-used-to-produce-them/
- https://www.sciencedirect.com/science/article/pii/S2162253124001100
- https://iubmb.onlinelibrary.wiley.com/doi/abs/10.1042/BA20030002
- https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0029101
- https://genome.cshlp.org/content/31/1/75
About The Author:
 Philip Probert, Ph.D., is a strategic scientific leader with extensive experience in bioprocessing and strategy development. He currently serves as the biologics technology lead at CPI, where he heads up the development department, delivering a diverse portfolio of innovation projects focused on the development and scale-up of biologic therapeutics and vaccines. His expertise spans all biologic modalities, with particular subject expertise in synthetic and cell-free manufacturing processes for nucleic acids and proteins. Probert holds a Ph.D. in hepatic toxicology from Newcastle University and an MBA from the Open University.
Philip Probert, Ph.D., is a strategic scientific leader with extensive experience in bioprocessing and strategy development. He currently serves as the biologics technology lead at CPI, where he heads up the development department, delivering a diverse portfolio of innovation projects focused on the development and scale-up of biologic therapeutics and vaccines. His expertise spans all biologic modalities, with particular subject expertise in synthetic and cell-free manufacturing processes for nucleic acids and proteins. Probert holds a Ph.D. in hepatic toxicology from Newcastle University and an MBA from the Open University.