
The Clinical Landscape Of ADCs In 2023: Diverse Technologies, Narrow Target
By Stuart D. Barnscher, ZymeWorks Inc.

Antibody-drug conjugates (ADCs) are a type of targeted cancer therapy that combines the specificity of monoclonal antibodies with the potency of cytotoxic drugs. This exciting modality is revolutionizing the treatment landscape for cancer by targeting specific proteins on the surface of cancer cells and delivering a toxic payload directly to the tumor resulting in enhanced efficacy compared to traditional chemotherapy. In recent years, there has been explosive growth in the development of ADCs. Between 2019 and 2022, eight ADCs were approved by the FDA, and in 2022, 57 new ADCs entered Phase 1 clinical trials, a 90% increase compared to 2021 (Figure 1). Additionally, 249 clinical trials evaluating ADCs were initiated in 2022, a 35% increase compared to 2021 (Figure 2).
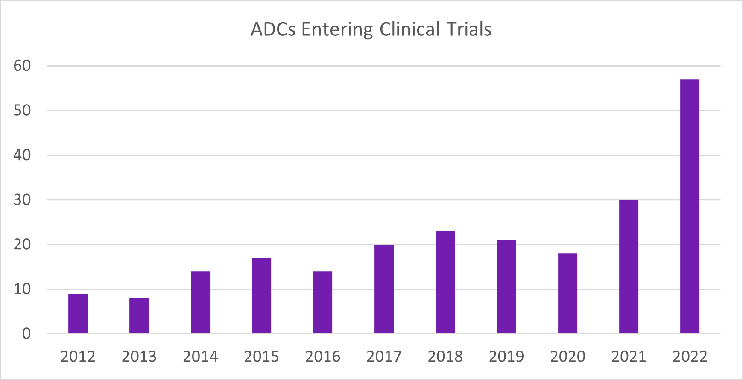
Figure 1. ADCs entering clinical trials per year from 2012 to 2022. Figure provided by and used with permission from Beacon Targeted Therapies by Hanson Wade.
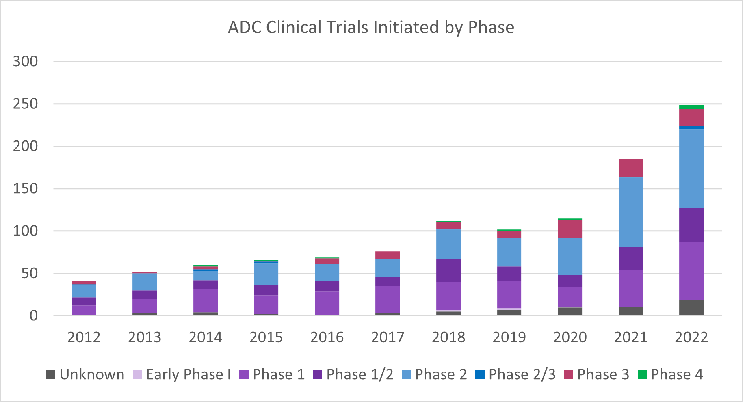
Figure 2. ADC clinical trials initiated by phase per year from 2012 to 2022. Figure provided by and used with permission from Beacon Targeted Therapies by Hanson Wade.
As drug development experts strive to discover innovative strategies that can offer maximum benefits in terms of drug targeting, safety, and efficacy, the widespread interest in ADCs has given rise to a paradoxical situation. On one hand, there is a notable overlap in the cancer targets being investigated, while on the other hand, there is an enormous variety in the ADC technologies utilized to develop each ADC. Currently, the two primary cancer targets being investigated for ADC development are HER2 and TROP2, which together make up 20% of all ADC programs in clinical development (Figure 3). It is worth noting that these targets remained the top two in terms of new program disclosures in 2022, accounting for 29% of the overall target space. Although some developers may utilize HER2 and TROP2 to establish proof of concept for their ADC technologies or ideas, it is disheartening to witness persistent investments in this crowded target space that may only result in marginal improvements for patients.
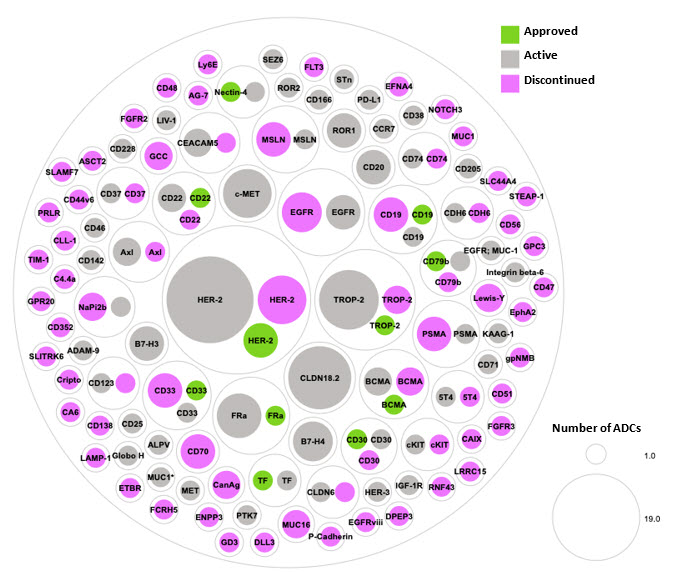
Figure 3. Approved, active, and discontinued clinical-stage ADC programs organized by target. Green represents an approved ADC, gray represents an ADC currently in clinical trials, and pink represents a discontinued ADC program.
Over the past two decades, significant investments have been made in every component of an ADC, including the antibody, conjugation, linker, and payload. This has led to the creation of a wide range of tools that can be used to build new drug conjugates. Conjugation methods include stochastic techniques that utilize covalent linkage to cell surface lysines or interchain cysteines after disulfide reduction. Site-specific conjugation methods have focused on generating highly stable and near homogeneous ADC products. This has been achieved using a variety of techniques including engineered cysteine mutations, sortase-mediated conjugation, enzymatic glycoengineering, intein-mediated conjugation, click chemistry, and non-natural amino acid-mediated conjugation, to name a few. Linker chemistry has seen significant innovation, with a large number of distinct linkers disclosed, 33 of which have been employed in clinical molecules. Novel payloads have also received substantial investment, with >60 distinct payloads used in clinical-stage ADCs. Despite the diversity of payload entities, the payload mechanism of action for most ADCs has been restricted, with 51% of ADCs in clinical development utilizing tubulin inhibitors. It is promising that only 25% of new ADCs disclosed in 2022 use tubulin inhibition as the payload mechanism of action, indicating an increase in the diversity of payload mechanism and the potential to better pair payload mechanism with tumor and target biology (Figure 4).

Figure 4. Proportion of ADCs stratified by payload mechanism of action. Outer doughnut represents the proportion of payload mechanism of action for all ADCs in clinical development. Inner doughnut represents the proportion of payload mechanism of action for newly disclosed ADCs in 2022. Figure provided by and used with permission from Beacon Targeted Therapies by Hanson Wade.
To illustrate the variety of ADC technologies employed in designing molecules, we will examine the design features and characteristics of ADCs targeting the most common ADC target, HER2. Despite being a well-established target, HER2 continues to be heavily researched due to its relevance in cancer treatment and clinical validation.
ADCs Against Human Epidermal Growth Factor Receptor 2 (HER2)
Human epidermal growth factor receptor 2 (HER2) is a cell surface receptor that is overexpressed in a variety of cancers, including breast, gastric, and other solid tumors. The overexpression of HER2 is associated with increased cancer cell proliferation, survival, and metastasis and has been extensively explored by researchers. Currently, there are two FDA approved HER-targeting ADCs: Kadcyla, approved in 2013, and Enhertu, approved in 2019. Disitamab vedotin is approved in China and under investigation in multiple Phase 2 trials worldwide. Table 1 highlights the key features of selected HER2 ADCs. There is a diverse range of ADC technologies and design principles used to construct these molecules. Kadcyla was the first HER2 ADC developed conjugating the maytansinoid payload DM1 to trastuzumab with an average DAR of 3.5. Enhertu utilizes an antibody with the same sequence of trastuzumab conjugated to the relatively low potency camptothecin payload, DXd, with a DAR of 8 and showed an improvement over Kadcyla in the head-to-head trial DESTINY-Breast03. A high DAR would not be possible for more potent microtuble inhibitor payloads in the auristatin class as seen with zanidatamab zovodotin, ARX788 (employing a DAR of 2) or disitamab vedotin (employing a DAR of 4). Similarly, trastuzumab duocarmazine, under regulatory review, utilizes a highly potent DNA alkylating duocarmycin payload conjugated to a DAR of 2.
A variety of conjugation methods have been used to control drug loading or improve stability (for example in ARX788 and MEDI4276); however, these efforts have not substantially improved the efficacy or safety profiles compared to ADCs employing stochastic conjugation methods. ZW49 and MEDI4276 utilize a biparatopic antibody able to recognize two distinct epitopes on HER2. This approach was unsuccessful for MEDI4276 due to severe toxicity at low doses, but it is challenging to determine if the main driver of toxicity was the tetravalent biparatopic antibody or if it was the use of an ultra-potent tubulysin payload conjugated to a DAR of 4 using a stable site-specific engineered cysteine conjugation technology. Out of the 42 HER2 ADCs that have undergone clinical trials, only seven have been mentioned in this discussion, but they exemplify the diversity of technological approaches that researchers have taken in an attempt to maximize efficacy and minimize toxicity.
Table 1. Comparison of design features and technologies used in seven HER2 ADCs
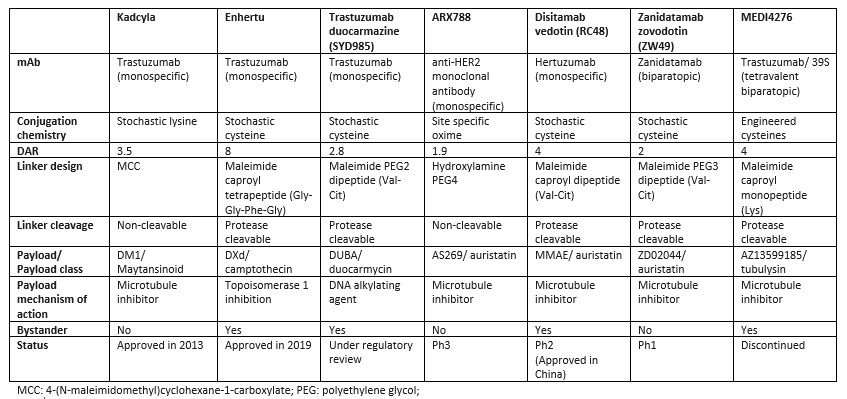
The diversity of approaches highlights the complexity of developing effective ADCs, as each component of the ADC can impact its overall efficacy and safety profile. It’s important to remember that each ADC is unique as researchers simultaneously modify various components, such as the antibody, conjugation chemistry, linker, and payload to achieve the desired results. This uniqueness creates challenges when dissecting which ADC design feature or component is driving the desired efficacy or undesired toxicity.
A silver lining in this challenging research environment is that the rapid expansion of interest in ADC technology is driving efforts to better understand how each ADC component can be optimized, not in isolation but as a part of an integrated design philosophy and in relation to clinical results. These insights can provide essential support and guidelines to improve efficiency, curtail toxicity, and reduce redundance in research in the years ahead.
About The Author:
 Stuart D. Barnscher leads the discovery and development of novel drug candidates and ADC technologies with a focus on antibody discovery and engineering, in vitro biology, and in vivo pharmacology and pharmacokinetics at Zymeworks. He has led multiple research programs and made significant contributions to the design and development of ZW49. He was instrumental in the development of multiple ADC drug-linker technologies developed at Zymeworks, including the topoisomerase-1 inhibitor platform, the ZymeLink auristatin platform, the ZymeLink Hemiasterlin platform, and the toll-like receptor 7 (TLR-7) agonist platform. Barnscher obtained his undergraduate degree in biochemistry from the University of British Columbia.
Stuart D. Barnscher leads the discovery and development of novel drug candidates and ADC technologies with a focus on antibody discovery and engineering, in vitro biology, and in vivo pharmacology and pharmacokinetics at Zymeworks. He has led multiple research programs and made significant contributions to the design and development of ZW49. He was instrumental in the development of multiple ADC drug-linker technologies developed at Zymeworks, including the topoisomerase-1 inhibitor platform, the ZymeLink auristatin platform, the ZymeLink Hemiasterlin platform, and the toll-like receptor 7 (TLR-7) agonist platform. Barnscher obtained his undergraduate degree in biochemistry from the University of British Columbia.