
The Matchmaker Modality: Engineering Event-Driven Pharmacology With Induced Proximity
By Mahmoud Khatib Al-Ruweidi

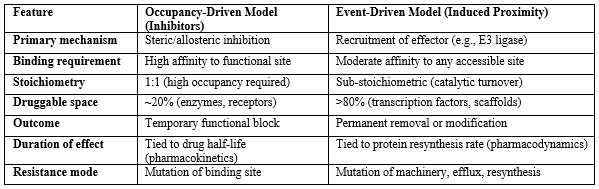
For decades we were taught a simple rule: a small molecule works only while it occupies its target. Induced proximity pharmacology breaks that rule by turning ligands into matchmakers that initiate biology (degradation, relocalization, stabilization) rather than merely blocking function. Clinical evidence recently confirmed that protein degraders can outshine conventional therapies. A recent trial on the estrogen receptor degrader vepdegestrant showed that it doubled the progression-free survival and lowered adverse events for ER-positive breast cancer patients compared to fulvestrant.1,2 This significant finding confirmed that a small molecule can catalyze protein elimination without continuously occupying its active site. Traditionally, medicinal chemistry focused on the occupancy model, where drugs targeted enzymes with accessible pockets, making many proteins "undruggable."3 Induced proximity counters this, using bifunctional compounds or engineered binders to bring target proteins near cellular effectors like E3 ubiquitin (Ub) ligases, initiating processes such as ubiquitination, endocytosis, or deubiquitination.4,5 Upon the trigger of the event, the drug can dissociate and catalyze the same reaction repeatedly, thereby decoupling efficacy from continuous occupancy.6 The success of vepdegestrant and the rapid diversification of induced proximity modalities suggest that event-driven pharmacology is poised to expand therapeutic reach beyond enzymes and receptors into transcription factors, scaffolding proteins, and signaling complexes.7
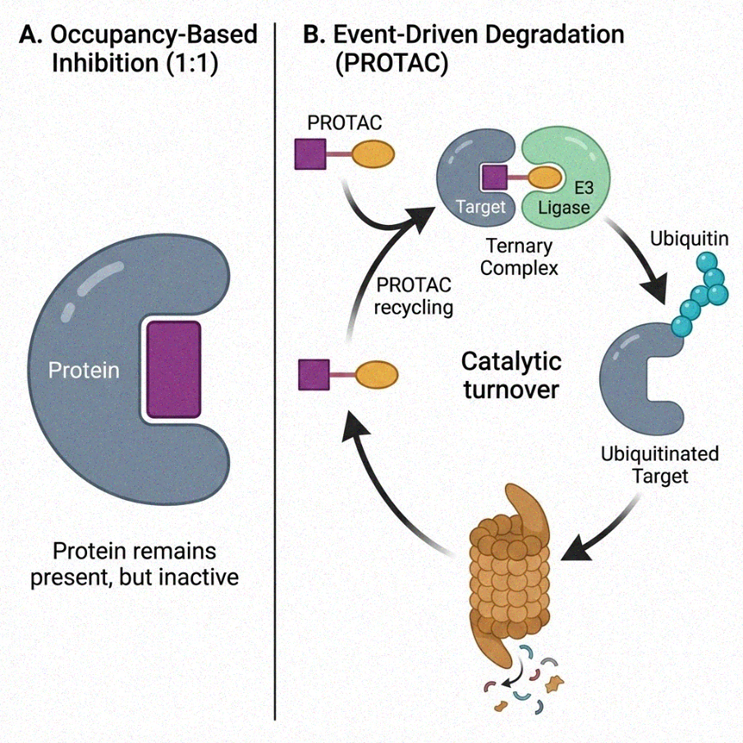
Figure 1. Paradigm shift from occupancy-based inhibition to catalytic, event-driven pharmacology.
(A) Occupancy model; (B) Event-driven model
Mechanistic Foundations: Ternary Complexes And Catalytic Turnover
The essence of any induced proximity strategy is the formation of a ternary complex that consists of the target protein, a recruited effector, and the bridging molecule.8 This three-body interaction varies fundamentally from a convenient bimolecular binding event because it exhibits cooperativity: binding of the effector can stabilize binding of the target (positive cooperativity) or interfere with it (negative cooperativity).9 Productive degraders often display slight positive cooperativity, making the ternary complex more stable than the binary pairs; negative cooperativity leads to weak complex formation and poor degradation. The degree of cooperativity is greatly sensitive to the geometry imparted by linkers connecting the ligands; subtle changes in linker length, rigidity, or orientation can convert a productive degrader into an inert one.
Emerging computational approaches, such as PRosettaC, leverage conformational flexibility to enhance the prediction of ternary complex structures and inform linker design, offering improvements over conventional AlphaFold-based methodologies.10 Machine learning and molecular dynamics simulations can further aid in navigating the immense design space by proposing linkers and warheads that maximize stability and cooperativity. However, a universally applicable algorithm for constructing the optimal complex does not currently exist; medicinal chemists continue to depend on iterative synthesis and screening to optimize cooperativity.
Another defining feature of induced proximity drugs is their catalytic, event-driven mode of action. Since the drug is not consumed in the process, one molecule can catalyze the modification or degradation of multiple target proteins in succession.6 For example, after recruiting a target for E3 ligase, the degrader dissociates once ubiquitination is initiated and can engage another target, enabling sub-stoichiometric dosing. For this catalytic turnover to be efficient, the ternary complex must persist long enough for the biochemical event to complete, but not so long that the drug becomes sequestered in an inactive complex. This introduces the concept of residence time: a complex that dissociates too slowly becomes occupancy-like, while one that falls apart too quickly may release the target before polyubiquitination. Biophysical techniques, such as the surface plasmon resonance and isothermal titration calorimetry, help measure the stability of ternary complexes and inform linker optimization; cell-based assays quantify degradation kinetics via metrics like DC50 (the degrader concentration causing 50% target depletion). The goal is to achieve a "Goldilocks" balance — enough stability for productive catalysis but sufficient dissociation for turnover.
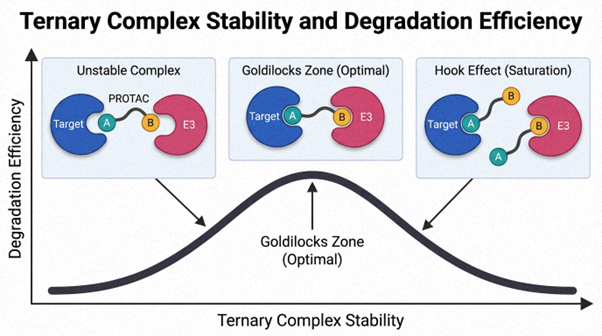
Figure 2. Relationship between ternary complex stability and degradation efficiency (“Goldilocks” behavior)
Designing Bifunctional Molecules: Target And Effector Recruitment
To create an effective degrader, a careful selection of three components is needed:
- A ligand that binds the protein of interest
- A ligand that recruits the effector (such as an E3 ligase or a deubiquitinase [DUB])
- A linker connecting them
The target-binding ligand does not necessarily need an extreme high affinity. In many cases, moderate affinity ligands have been converted into potent degraders due to cooperativity within the ternary complex compensating for weaker binding.8 Nevertheless, high specificity remains vital to avoid dragging the wrong protein into the degradation machinery and resulting in an off-target toxicity. At the effector end, early degrader programs relied almost exclusively on ligands for two well-studied E3 ligases, cereblon (CRBN) and von Hippel–Lindau (VHL).11,12 These ligases functioned as generic handles to recruit the proteasome. The human genome encodes more than 600 E3 ligases, plenty with tissue-specific expression, prompting intensive efforts to discover ligands for alternative ligases, such as IAP, MDM2, DCAF1, DCAF16, and KLHDC2.13 Covalent recruiters targeting less-explored ligases, such as RNF114 and FEM1B, have been developed through reactive fragments and natural product derivatives such as nimbolide.14 Expanding the recruiter repertoire allows designers to match the E3 ligase to the biology of a disease — for instance, choosing a ligase highly expressed in cancer cells but absent in normal tissues or using multiple ligases to counteract resistance.
The linker connecting the two ligands is more than a passive tether; its length, flexibility, and physicochemical properties profoundly influence ternary complex formation and cell permeability.15 Rigid linkers promote beneficial geometries but may hinder permeability, while flexible linkers raise entropic costs yet enable conformational sampling. Linker optimization methods include empirical scanning, iterative modification, and structure-guided strategies. Reducing polar surface area or introducing intramolecular hydrogen bonds can enhance cell uptake and oral bioavailability for large degrader molecules (700 to 1,200 Da).
Chemists draw on diverse chemical spaces to find both target warheads and recruiter ligands. Fragment-based drug discovery, DNA-encoded libraries, and natural product mining all contribute to the expanding toolbox of binding motifs. Natural products have historically provided potent ligands for E3 ligases (e.g., derivatives of thalidomide for CRBN or nimbolide for RNF114).16 Modern synthetic chemistry provides bespoke reactive fragments that covalently capture shallow pockets on lesser known ligases or deubiquitinases (DUBs), while encoded libraries enable high-throughput screening of millions of macrocyclic peptides and small molecules. The convergence of synthetic and natural chemistry underscores the multidisciplinary nature of induced proximity discovery: chemists must balance potency, selectivity, covalency, and drug-like properties while considering how a ligand's origin influences patentability and development cost.
Beyond Proteasomal Disposal: Lysosomal And Autophagic Routes
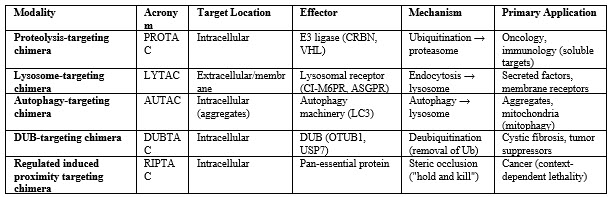
The induced proximity concept is not limited to proteasomal degradation. Lysosome-targeting chimeras (LYTACs) couple extracellular or membrane proteins to receptors like CI-M6PR that direct cargo to lysosomes, enabling removal of secreted proteins or cell-surface receptors that PROTACs cannot reach.17 Synthetic "EndoTags" fused to antibodies or binding proteins are capable of triggering internalization of cell-surface proteins followed by lysosomal trafficking. Autophagy-targeting chimeras (AUTACs) use a degradation tag recognized by autophagy receptors to mark organelles, protein aggregates, or pathogens for clearance through the autophagosomes.18 AUTACs act independently of ubiquitination, providing options when proteasomal pathways are impaired. Variants, such as autophagosome tethering compounds (ATTECs) and autophagy-targeting chimera (AUTOTAC) molecules, recruit other autophagy receptors or E3 ligases to direct specific cargoes for autophagic removal, broadening therapeutic applications.19
These lysosomal and autophagy-based strategies are especially promising for conditions characterized by toxic extracellular proteins or large intracellular structures. LYTACs could eliminate circulating pathogenic proteins, including antibodies or hormones, and modulate cell-surface receptors implicated in cardiovascular or metabolic diseases. AUTACs offer a way to remove protein aggregates or dysfunctional mitochondria implicated in neurodegenerative disorders or inborn errors of metabolism.18 Proof-of-concept studies have shown that compounds like AUTAC4 can clear defective mitochondria in cells derived from Down syndrome patients and improve mitochondrial function.18 Although these approaches face challenges in delivery and immunogenicity (such as antibody conjugates and many LYTACs are large proteins), they showcase how induced proximity can harness different cellular disposal systems beyond the proteasome.
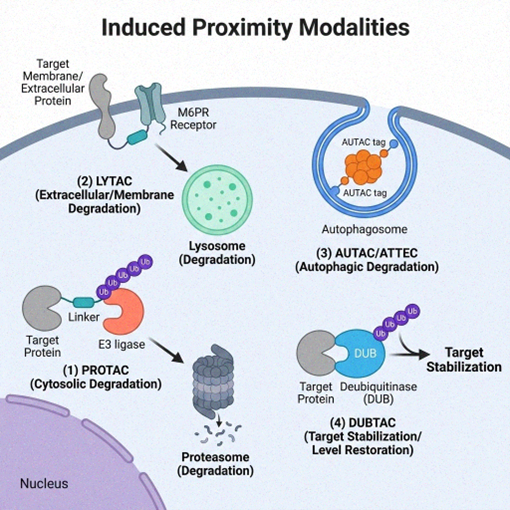
Figure 3. Diversity of induced proximity technologies for targeted protein modulation
Stabilization Via DUBTACS And Modulation Of Protein Function
Induced proximity can be reversed to stabilize proteins that are underexpressed or degrade rapidly. DUB-targeting chimeras (DUBTACs) bring DUBs to target proteins, removing Ub chains and preventing degradation. This may help diseases from loss-of-function mutations by extending protein half-life instead of relying on gene therapy. The first PRO-DUBTACs used VHL and ovarian tumor domain deubiquitinase, ubiquitin aldehyde–binding 1 (OTUB1) ligands to stabilize tumor suppressors and inhibit tumor growth in vitro.20 For example, DUBTACs could preserve mutated p53 activity by blocking its degradation. However, developing selective DUB ligands is challenging due to limited availability, and balancing ligase and DUB activities requires careful design. Successful stabilization must restore function, as accumulated misfolded proteins are ineffective. Overall, DUBTACs broaden induced proximity approaches to regulate protein homeostasis.

Regulated Induced Proximity And Targeted Cell Killing
A more recent innovation, the regulated induced proximity targeting chimera (RIPTAC), exploits the paradigm for selective cell killing rather than protein elimination.21 A RIPTAC brings an essential "housekeeping" protein into proximity with a tumor-specific protein, forming a tri-complex that sequesters or inactivates the essential protein only in cells expressing the tumor marker.21 This "hold-and-kill" concept offers exquisite selectivity: healthy cells lacking the tumor marker do not form the ternary complex and the essential protein remains active. Early preclinical compounds (H001, H003) targeting the androgen receptor (AR) and an undisclosed essential protein induced potent antiproliferative effects in AR-positive prostate cancer cells, and Halda Therapeutics' HLD-0915 entered Phase 1/2 trials as the first clinical RIPTAC.22 RIPTACs can bypass resistance from target mutations since they do not need strong binding to either partner. Their design focuses on positive cooperativity, benign individual ligands, and sufficient cell penetration despite their large size. If successful, RIPTACs may advance precision oncology and other fields requiring targeted cell elimination.
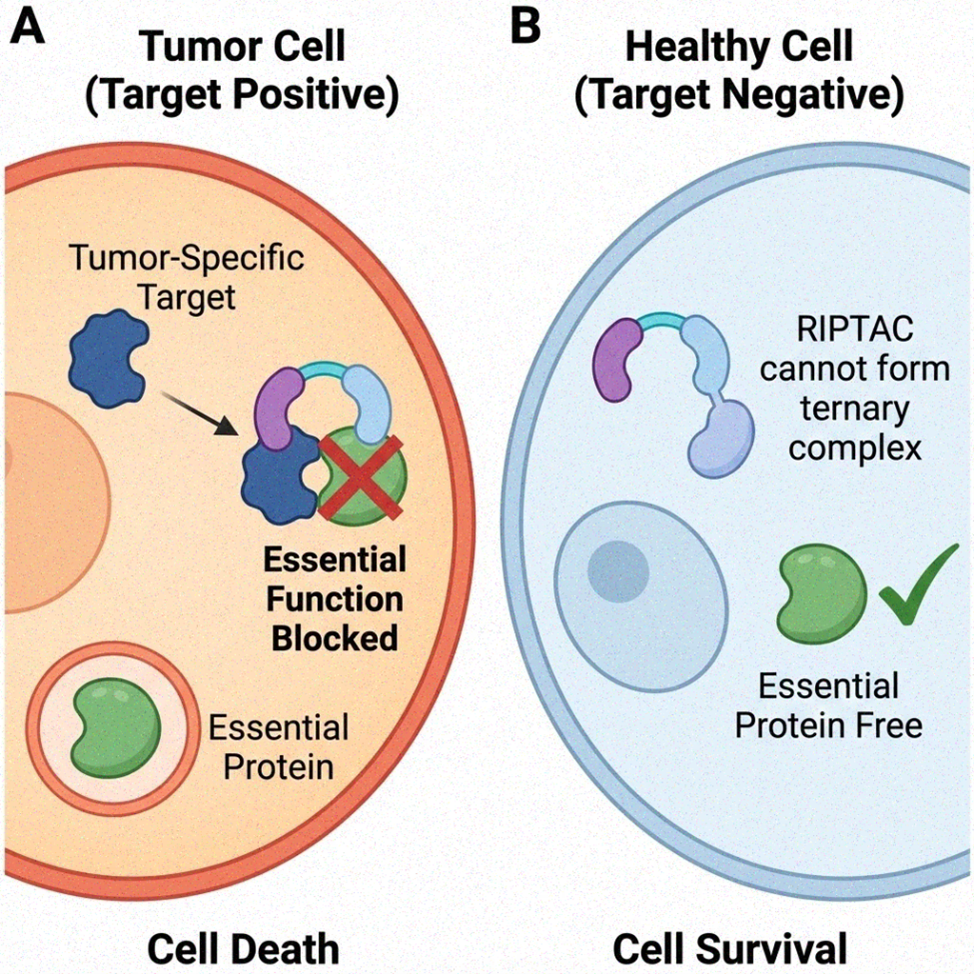
Figure 4. Selective cell killing by RIPTACs through functional sequestration. (A) Tumor cell; (B) Healthy cell
Challenges: Resistance, Pharmacology, And Development Hurdles
Despite remarkable promise, induced proximity therapeutics face substantial challenges. Resistance mechanisms that plague classical inhibitors also apply here. Cancer cells can down regulate or mutate the recruited effector (e.g., CRBN or VHL) so that the degrader cannot form the ternary complex.23 Upregulation of drug efflux pumps, such as P-glycoprotein, can reduce intracellular concentrations of large bifunctional molecules, hampering efficacy.24 Mutations or splice variants in the target protein may reduce binding affinity; though cooperativity can buffer modest affinity reductions, significant alterations can abolish degrader binding. Cells also can reroute the ubiquitination machinery by upregulating compensatory ligases or altering Ub pathway components.
- Pharmacokinetics and physicochemical properties present challenges: degraders often have high molecular weight and polarity, limiting oral bioavailability and tissue penetration. Many are subject to efflux or instability from metabolic enzymes and DUBs. Solutions include optimizing linkers, masking polar groups with intramolecular hydrogen bonds, prodrug strategies, and using antibody or nanoparticle conjugates — though these may increase immunogenicity and manufacturing complexity.
- Another challenge is off-target activity. A bifunctional molecule may inadvertently bring an unintended protein into proximity with the effector, causing unwanted degradation or stabilization. Comprehensive proteomic profiling of degraders is therefore essential. Proteomics methods, such as tandem mass tag labeling combined with mass spectrometry, can quantify global protein abundance changes upon degrader treatment, revealing both on-target and off-target effects.25 Quantitative proteomics has already shown that some PROTACs degrade off-target proteins with surprising efficiency, underscoring the need for rigorous selectivity assessment.
- Regulatory and manufacturing issues are significant. Many degraders are large and complex, making them difficult to produce with standard small molecule methods. Agencies are still developing guidelines for classifying and evaluating these molecules, especially those like LYTACs that have protein components. Concerns about immunogenicity, biodistribution, and long-term safety remain unresolved. Developers also face a crowded patent landscape around E3 ligase recruiters and linker chemistries, requiring careful IP management.
Final Word: Rewriting Pharmacology
Induced proximity has evolved into a versatile platform with the potential to change drug discovery by enabling the degradation, stabilization, or modulation of previously undruggable proteins through ternary complexes. Successes such as vepdegestrant highlight the effectiveness of catalytic degraders, while innovations like LYTACs, AUTACs, DUBTACs, and RIPTACs show the approach’s adaptability. However, challenges remain in predicting suitable targets, understanding resistance, and ensuring safety. Progress will rely on collaboration across chemistry, biology, immunology, and computational science. Induced proximity shifts pharmacology toward managing protein fate rather than just blocking activity, opening new possibilities for therapeutic precision.
References
- Hamilton, E.P., Ma, C., De Laurentiis, M. et al. VERITAC-2: a Phase III study of vepdegestrant, a PROTAC ER degrader, versus fulvestrant in ER+/HER2- advanced breast cancer. Future Oncol. 20, 2447–2455 (2024).
- Gough, S.M., Flanagan, J.J., Teh, J. et al. Oral Estrogen Receptor PROTAC Vepdegestrant (ARV-471) Is Highly Efficacious as Monotherapy and in Combination with CDK4/6 or PI3K/mTOR Pathway Inhibitors in Preclinical ER+ Breast Cancer Models. Clin. Cancer Res. 30, 3549–3563 (2024).
- Crews, C.M. Targeting the undruggable proteome: the small molecules of my dreams. Chem. Biol. 17, 551–555 (2010).
- Sakamoto, K.M., Kim, K.B., Kumagai, A., Mercurio, F., Crews, C.M. & Deshaies, R.J. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 98, 8554–8559 (2001).
- Békés, M., Langley, D.R. & Crews, C.M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
- Bondeson, D.P., Mares, A., Smith, I.E.D. et al. Catalytic in vivo protein knockdown by small molecule PROTACs. Nat. Chem. Biol. 11, 611–617 (2015).
- Lai, A.C. & Crews, C.M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16, 101–114 (2017).
- Gadd, M.S., Testa, A., Lucas, X. et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 13, 514–521 (2017).
- Roy, M.J., Winkler, S., Hughes, S.J. et al. SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 14, 361–368 (2019).
- Zaidman, D., Prilusky, J. & London, N. PRosettaC: Rosetta Based Modeling of PROTAC Mediated Ternary Complexes. J. Chem. Inf. Model. 60, 4894–4903 (2020).
- Buckley, D.L., Van Molle, I., Gareiss, P.C. et al. Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules to Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 134, 4465–4468 (2012).
- Ito, T., Ando, H., Suzuki, T. et al. Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 (2010).
- Li, W., Bengtson, M.H., Ulbrich, A. et al. Genome-Wide and Functional Annotation of Human E3 Ubiquitin Ligases Identifies MULAN, a Mitochondrial E3 that Regulates the Organelle's Dynamics and Signaling. PLoS ONE 3, e1487 (2008).
- Spradlin, J.N., Hu, X., Ward, C.C. et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 15, 747–755 (2019).
- Cyrus, K., Wehenkel, M., Choi, E.Y., Lee, H., Swanson, H. & Kim, K.B. Impact of linker length on the activity of PROTACs. Mol. BioSyst. 7, 359–364 (2011).
- Fischer, E.S., Böhm, K., Lydeard, J.R. et al. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014).
- Banik, S.M., Pedram, K., Wisnovsky, S., Ahn, G., Riley, N.M. & Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020).
- 18. Takahashi, D., Moriyama, J., Nakamura, T. et al. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 76, 797–810.e10 (2019).
- 19. Ji, C.H., Kim, H.Y., Lee, M.J. et al. The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nat. Commun. 13, 904 (2022).
- Henning, N.J., Boike, L., Spradlin, J.N. et al. Deubiquitinase-targeting chimeras for targeted protein stabilization. Nat. Chem. Biol. 18, 412–421 (2022).
- Raina, K., Forbes, C.D., Stronk, R. et al. Regulated induced proximity targeting chimeras—RIPTACs—A heterobifunctional small molecule strategy for cancer selective therapies. Cell Chem. Biol. 31, 1490–1502.e42 (2024).
- ClinicalTrials.gov Identifier: NCT06800313. A Study of HLD-0915 in Patients With Metastatic Castration Resistant Prostate Cancer (mCRPC). First patient dosed February 24, 2025.
- Zhang, L., Riley-Gillis, B., Vijay, P. & Sher, Y. Acquired Resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes. Mol. Cancer Ther. 18, 1302–1311 (2019).
- Kurimchak, A.M., Herrera-Montávez, C., Montserrat-Sangrà, S. et al. The drug efflux pump MDR1 promotes intrinsic and acquired resistance to PROTACs in cancer cells. Sci. Signal. 15, eabn2707 (2022).
- Donovan, K.A., Ferguson, F.M., Schieber, J.W. et al. Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development. Cell 183, 1714–1731.e10 (2020).
About The Author
 Mahmoud K. Al-Ruweidi is a pharmaceutics specialist with expertise in rational drug design, discovery, and delivery. Trained as a biomedical engineer, his research spans formulation science and bioengineering approaches to medicine. He has worked across biochemistry, medical devices, and biomaterials, applying interdisciplinary methods to accelerate therapeutic innovation. Beyond the lab, he is an advocate for improving academic systems to better support young scientists and safeguard research integrity.
Mahmoud K. Al-Ruweidi is a pharmaceutics specialist with expertise in rational drug design, discovery, and delivery. Trained as a biomedical engineer, his research spans formulation science and bioengineering approaches to medicine. He has worked across biochemistry, medical devices, and biomaterials, applying interdisciplinary methods to accelerate therapeutic innovation. Beyond the lab, he is an advocate for improving academic systems to better support young scientists and safeguard research integrity.