
Harness High-Quality PBPK Modeling To Enhance Preclinical And Clinical Readiness
By Deanna Mudie, Ph.D., Senior Principal Engineer, Lonza Small Molecules, and John DiBella, President, SLP Division, Simulations Plus

Rapid and efficient development of drug candidates sits top of mind for pharmaceutical companies with funding constraints looking to accelerate timelines. Unfortunately, many early drug candidates demonstrate poor oral absorption properties, which can make it challenging to achieve target pharmacokinetic profiles. Without up-front knowledge of absorption risks and mitigation strategies, poor absorption can significantly impact preclinical and clinical study timelines and costs.
However, advancements in physiologically based pharmacokinetic (PBPK) modeling are making it easier than ever to identify and mitigate absorption risks in early drug development. A wide range of approved drugs have utilized PBPK modeling to develop safer, more effective formulations and eliminate unnecessary animal and human studies. Utilizing PBPK modeling in your early development studies can help you to identify drug absorption risks, formulate risk mitigation strategies, and de-risk your preclinical and clinical study outcomes.
What is PBPK Modeling?
PBPK models represent animals and humans as virtual collections of organs and tissues, each defined by a system of mathematical equations. They are developed using quantitative values that describe characteristics like body weight, blood flow rate, physicochemical properties, and formulation, as well as mechanisms like dissolution, precipitation, absorption, and metabolism. A well-honed set of parameters describing drug properties, formulation information, metabolism and excretion properties, as well as a description of the underlying system physiology to be modeled, is used to simulate and predict the local exposure of a drug at the site of administration. As it is absorbed into the bloodstream, the model demonstrates how it is distributed and eliminated around the body.
Over the past 20 years, PBPK modeling has increasingly shown value for clinical trial design. The early information collected in animals can be used to help extrapolate expectations for in-human clinical trials. From there, the clinical data collected in specific human populations can be extrapolated for other subgroups, including pediatrics, certain disease populations, and groups where it may be difficult to design a clinical study due to ethical considerations.
The Current Climate of PBPK Modeling
The relationship between PBPK modeling and research and discovery (R&D) continues to evolve. When Lonza first started collaborating with drug sponsors to discuss PBPK goals, there was a significant amount of skepticism. It was difficult to comprehend how all the different biological, chemical, and physical processes that occur in vivo could be described through a set of mathematical equations. Early adopters were interested in how modeling and simulation could make predictions to support their studies. As confidence in these approaches increased, stakeholders no longer needed to be convinced of PBPK modeling’s value. Instead, clients began to ask how they could use these approaches to enhance their development program and make downstream decisions.
Presently, we’ve reached a stage where many companies around the world appreciate the value of modeling, simulation, and PBPK. They are beginning to reevaluate their internal standard operating procedures (SOPs) and R&D process to reflect the availability of PBPK approaches. Some are trying to understand how early they can incorporate PBPK across their therapeutic programs to inform which experiments to run and studies to perform. Modeling and simulation alongside experiments can reduce costs and accelerate timelines to market. Recently, there has been a substantial rise in the number of companies recruiting scientists with experience in PBPK modeling, not only in large pharma but in companies of all shapes and sizes across different markets.
Five years ago, the Food and Drug Administration (FDA) and European Medicines Agency (EMA) began to publish guidance documents on PBPK modeling, including how to frame results to support regulatory interactions. These guidance documents are highly specific and explain how to apply PBPK in drug development programs. As a result, there has been a significant rise in the number of submissions that used PBPK modeling to inform a regulatory decision or in lieu of running clinical studies. Two recent publications from the FDA, one from the Office of Clinical Pharmacology and one from the division of Biopharmaceuticals in the Office of New Drug Products, summarize the submissions they have received over a certain period where PBPK modeling was referenced. In Figure 1 on the left, the graph demonstrates the diversity of PBPK applications to support clinical pharmacology applications. It can be used to create predictions in special populations and support drug-drug interaction evaluations, as well as assess the impact of food.
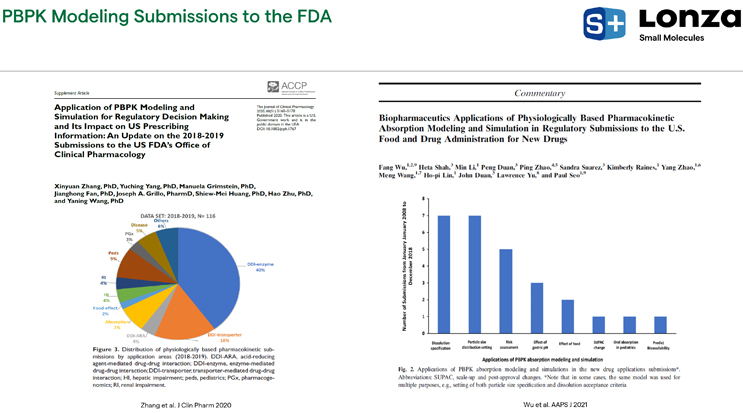
Figure 1. Recent FDA publications on PBPK modeling
In Figure 1 on the right, the graph shows ways companies utilize PBPK to help support biopharmaceutical regulatory interactions. This tool can be used in early development while looking for a better understanding of which direction a program should move in and can continue through to post-approval. Once a drug is on the market, PBPK modeling can provide insight into manufacturing process changes.

Figure 2. Pfizer study on in vitro-in vivo extrapolation using PBPK
In the early development stage, PBPK modeling has been validated to support dose selection and study design for first-in-human trials. Pfizer research looked at over 20 of their own compounds that were in clinical development to quantify how well they could predict first-in-human exposure using PBPK in an in vitro-in vivo extrapolation (IVIVE) approach versus traditional allometric scaling, which relies only on animal in vivo data before scaling up to the human environment (Figure 2). The takeaway was that when using in vitro information as input to predict human IV or oral PK outcomes, PBPK approaches do as well or better in all cases than allometric scaling.
Over the years, more publications have been released, leading to updated model-building strategies to help support in-human predictions. Many studies include suggestions on how scientists in different stages of projects could start using PBPK to help make better informed decisions. Roche published useful guides almost 20 years ago that outlined their strategies for incorporating PBPK modeling as a standard procedure for all programs, including a chart outlining when modeling and simulation were being used in preclinical development to help select clinical formulation concepts for their first-in-human studies (Figure 3). In a publication from 2006, Roche examined a case study for a BCS class II drug, where, based upon the Biopharmaceutics Classification System (BCS)assessment, there were suggestions internally to utilize solubility enhancement approaches to improve the oral bioavailability of this drug in dogs (Figure 3).

Figure 3. Roche PBPK modeling approach and study
Roche was making a significant investment in a formulation technology to try to get more drug absorbed into the systemic circulation. Before they went down that path, they performed parameter sensitivity analysis (PSA) to assess the impact of changes to certain formulation or drug characteristics on the absorption and bioavailability of the drug. The early PSA runs showed that, surprisingly, changes to particle size or solubility by technological means would not necessarily lead to improved bioavailability (Figure 4). There wasn’t a big change in bioavailability due to changes in particle size unless the particles were large (> 50 microns); likewise, there wasn’t a major change in the bioavailability due to changes in solubility unless the solubility was significantly reduced.
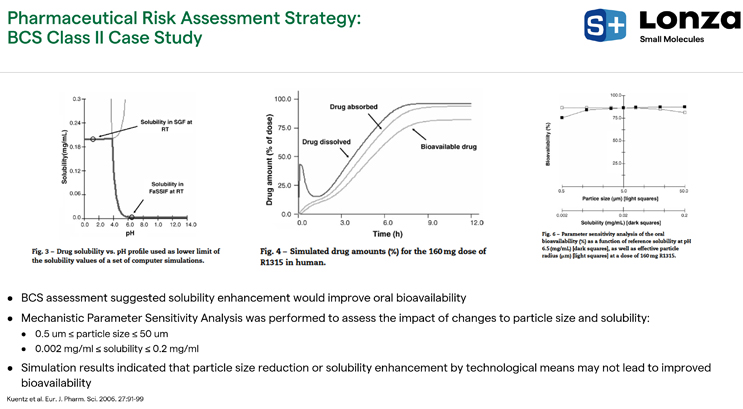
Figure 4. BCS Class II case study findings
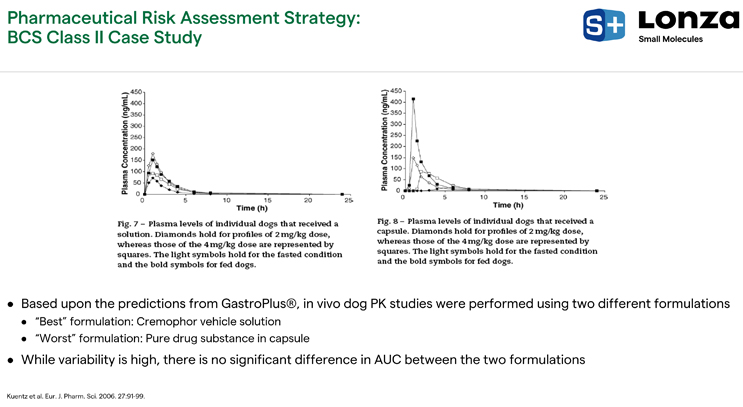
Figure 5. GastroPlus® predictions for BCS Class II case study
Roche validated what they saw from these analyses and went into dog studies where they developed multiple formulations. The “worst formulation” was pure drug substance in the capsule, demonstrated in the Figure 5 plot on the right. The “best formulation” was the Cremaphor vehicle solution where they tried, as best as possible, to put the drug in solution to improve the bioavailability (Figure 5, left). When accounting for variability effects, the area under the curves (AUCs) were similar between these formulations. There wasn't any added benefit of the vehicle solution path, which reflects the results of the PSA.
Oral Absorption Risks & Mitigation Strategies
Solubility and permeability are the key underlying mechanisms of poor oral absorption. BCS II and IV compounds have poor solubility in gastrointestinal fluids at (at least) one of their prescribed dosages, and BCS III and IV compounds have poor permeability (Figure 6). Compounds with poor solubility make up a large fraction of drug candidates in development, ranging from 70% and higher (Figure 6).
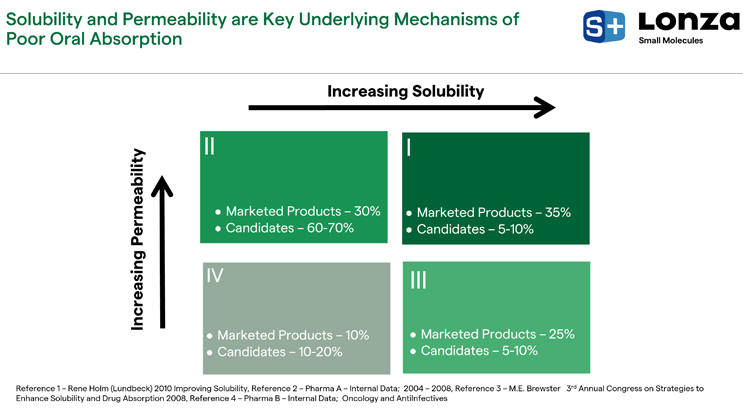
Figure 6. Solubility and permeability in marketed products and candidates
Solubility and permeability depend on a drug’s physical and chemical properties and gastrointestinal physiology. Key drug properties include lipophilicity, acid-base character and pKa, and melting point, as well as physiological properties like fluid pH and composition, fluid volumes, and fluid hydrodynamics. These properties, combined with the dose and formulation characteristics, drive absorption into the bloodstream. With in vitro testing and PBPK modeling, these properties can be varied with simulations and PSAs assessing the impact they’ll have on drug exposure.
There are several negative impacts that can arise from poor solubility and permeability in vivo. Low bioavailability is the key risk; in this instance, a preclinical or human subject might receive a dose and researchers would see a low fraction of said dose absorbed into the bloodstream. This can lead to failed preclinical or clinical studies because we're not able to assess toxicity or efficacy due to low exposure. Another prospective impact is an acid reducing agent (ARA) effect. There may be good exposure in the bloodstream without an ARA but with an ARA present, there may be diminished exposure with pH effects with weak base drugs.
Food effects are also important to look for. There may be higher exposure in the case of a positive food effect, or vice versa. ARA and food effects can lead to poor patient compliance and reduced efficacy if patients must follow complicated dosing regimens. In many cases, risks are diminished through formulation selection, which can often mitigate low bioavailability. We could see high plasma exposure with formulation strategies that are resistant to ARA and food effects, allowing for similar exposure in all cases and hopefully avoidance food labels or other restrictions.
There are several formulation strategies out there for mitigating poor oral absorption through solubility enhancement. Solid state alteration can be done via form or particle size. With this technique, you can select a different polymorph, create an amorphous solid dispersion, or micronize a drug. Other options include new crystalline compounds, perhaps cocrystals or salt forms of the API. Another option is solvation or complexation, where additional excipients, such as cosolvents, surfactants, cyclodextrins, or lipids are included in the dosage forms. The choice of the right technology for a given API depends on several factors, including the magnitude of the solubility enhancement needed, the dose, and the target product profile. In vitro testing and PBPK modeling can steer us in the right direction.
PBPK Modeling Services
Lonza’s PBPK modeling service offers three key outputs:
- Identify drug absorption risks: Early in the process we identify risks, including solubility, dissolution rate permeability, food effects, or pH dependent ARA effects.
- Recommend absorption risk strategies: If an absorption risk is identified, we recommend absorption risk mitigation strategies, including targeting a salt form, a cocrystal, or an amorphous solid dispersion, for example.
- Inform and de-risk preclinical or clinical study outcomes: Once we have formulations or drug products, we can start to inform or de-risk preclinical or clinical study outcomes by projecting the impact of dose, formulation characteristics, and food or pH dependent ARA effects. Our goal is to avoid unwanted surprises that can occur with poor absorption and to streamline the development process to get drugs to market faster.
Our service offerings include three components. First is the established ADMET Predictor® and GastroPlus® modeling software, which is validated and prevalent across the industry. Lonza also offers an expansive set of custom and off-the-shelf in vitro performance tests that are powerful for generating those inputs into the PBPK models. Lonza has vast experience with API synthesis, solubility enhancement, and formulation development, including having worked on over 10 amorphous solid dispersions that have progressed to the market, and over 20 patent families in amorphous or spray dry dispersion technology. In 2022, Lonza assessed 230 therapies in clinical development and supported about 140 commercial-scale small molecule projects.
Custom and off-the-shelf in vitro bioperformance tests are designed to be material sparing, using milligram quantities of API, and high throughput (Figure 7). Our custom amorphous solubility test measures amorphous solubility and precipitation prior to making a formulation. Pion® microDISS apparatuses evaluate factors such as dissolution and precipitation rates as well as drug speciation of drug product intermediates; custom inserts for our Pion® microDISS allow us to assess membrane flux to verify the impact of different dissolved drug species on diffusion. Furthermore, in-house controlled transfer dissolution tests allow us to determine the dynamic pH and transit impacts on dissolution and precipitation as a drug moves from the stomach to the small intestine.

Figure 7. Lonza’s suite of custom and off-the-shelf tools
PBPK serves as a bridge between drug substance and drug product and can be used in conjunction with solid form and particle engineering services to identify technologies and formulations and to develop and manufacture drug products for clinical trials and the market. PBPK can be extended throughout drug development, but the following case study focuses on the preclinical space leading up to Phase 1.
Case Study: Early API Absorption Risk Assessment
This case study emphasizes the impact of early API absorption risk assessments prior to obtaining preclinical data. In this exercise, we conducted an early API absorption risk assessment using model drug posaconazole. Posaconazole is an antifungal agent whose brand name is NOXAFIL®. It has oral dosages of 100 to 400 milligrams per administration and is dosed multiple times per day. Our inputs for this exercise were the API structure and in vitro crystalline and amorphous solubilities (Figure 8). That information was placed into the ADMET Predictor® and GastroPlus® with an in-house calculator to predict barriers to absorption, percent dose absorbed, and potential food or ARA effects.
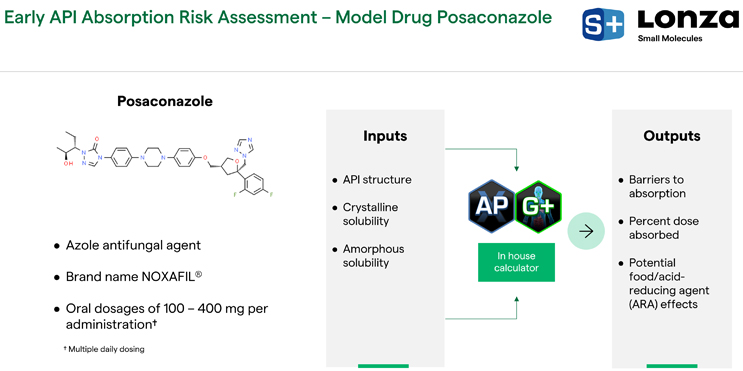
Figure 8. Posaconazole’s API structure, inputs, and outputs
Based on structure and solubility, we learned that posaconazole is a lipophilic weak base with solubility limited absorption. In Figure 9, the left plot demonstrates that solubility decreased as a function of pH, which drives a potential for precipitation as this drug moves from the stomach into the small intestine. It also has a high amount of bile salt micelle partitioning; the endogenous bile salt micelles in the gastrointestinal tract act to solubilize the drug. There was a 12-fold enhancement in solubility with 6.7 millimolar of bile salts in the media, which is representative of fasted state media. Per the developability classification system, posaconazole has a relatively high permeation rate, but it also has a high ratio of dose to solubility (Figure 9). Since it is over the dose range of 5 to 500 milligrams, it’s in the solubility limited regime and solubility is a barrier to absorption.
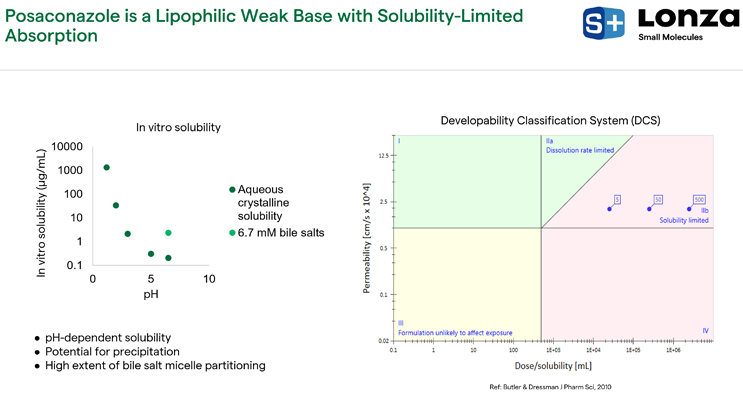
Figure 9. Posaconazole solubility findings
Next, PSAs were conducted using GastroPlus®, simulating a fasted human physiology, an immediate release tablet with relatively small particle size, and the Simulations Plus predicted permeation rate (S+Peff = 2 X 10-4cm/s). In Figure 10 on the lower left side, the simulated percent absorbed is relatively low: 25% absorption at doses greater than 100 milligrams. Therefore, we flagged an absorption risk for low bioavailability. On the right-hand side of Figure 10, gastric pH and precipitation of posaconazole when it enters the small intestine are both crucial factors due to large impact on simulated percent absorbed. When gastric pH is increased, there is a significant reduction in the percentage absorbed. However, when we simulate a relatively long precipitation time and achieve super saturation for a long duration, that impacts absorption as well. Overall, the percentage absorbed was in the range of 2% to 50%, which adequately predicts the behavior of the marketed NOXAFIL® oral suspension (OS) of posaconazole, which has an estimated bioavailability around 8% to 47% over the prescribed dose range. Posaconazole is believed to have a relatively low first pass extraction, and therefore bioavailability is indicative of the percent absorbed. By using the API structure and the measured crystalline solubility profile alone, these outcomes were well predicted.
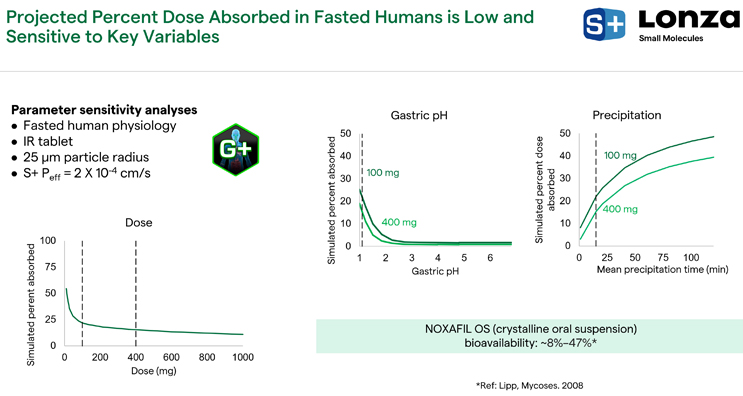
Figure 10. GastroPlus® PSA findings
We considered the potential for posaconazole to have pH-dependent drug interactions with ARAs and food. The left graph of Figure 11 demonstrates simulated percent absorbed as a function of gastric pH at a 100-milligram dose. When patients are taking ARAs, their gastric pH ranges from about 3 to 7; in this pH range, the simulated percent absorbed is significantly reduced compared to the fasted state. Data for NOXAFIL OS® indeed shows that exposure is reduced by 30% with an ARA, demonstrating a pH-dependent drug interaction with this molecule, demonstrating the validity of these predictions.
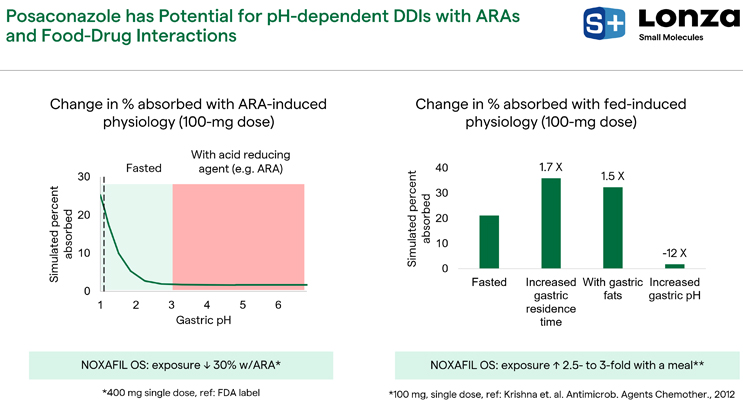
Figure 11. Posaconazole and pH-dependent drug interactions with ARAs and food
The right graph of Figure 11 shows what happens with fed-induced physiology changes for a 100-milligram dose, which flags the possibility of food effects. This evaluation was conducted by systematically changing three of the important variables that are affected with fed-state physiology. With an increased gastric residence time of about 3 hours, there is a 1.7-fold enhancement in percent absorbed. When we simulate fats in the stomach representative of a high fat meal, there is a 1.5-fold increase in simulated percent absorbed. With increased pH, which happens typically with a meal, there is a reduction in simulated percent absorbed. However, when people consume a meal, even though their pH might initially increase, the stomach experiences reacidification on the order of 1 to 4 hours. That can counteract reduced solubility of posaconazole during the residence time of the drug in the stomach. These simulations suggest that posaconazole has a potential sensitivity to food effects. These results are backed up by data for NOXAFIL OS®, which shows that when patients consume a meal, there is a 2.5- to three-fold increase in percent absorbed with a 100-milligram dose.
Given the predicted absorption risks highlighted above, including poor oral absorption in the fasted state and the potential for gastric pH-dependent ARA and food effects, we investigated mitigation strategies. We considered using an amorphous form of posaconazole and opted to use our custom in vitro solvent shift UV assay to measure the posaconazole amorphous solubility. We found a 30-fold enhancement in the amorphous compared to the crystalline solubility in intestinal media with bile salts representative of fasted humans. This prompted us to evaluate the amorphous form as a possible strategy for solubility enhancement and improved absorption. We did another set of PSAs using the same GastroPlus® model. In the lower left-hand corner of Figure 12, the graph shows that simulated percent absorbed is substantially increased using an amorphous form over a relatively large dose range.
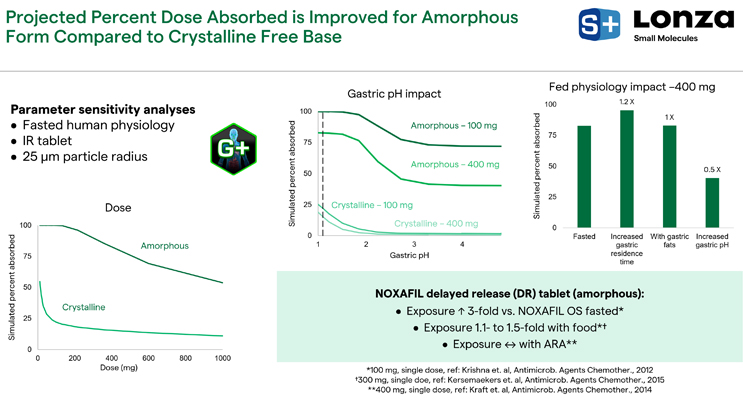
Figure 12. Amorphous form absorption projections
In the middle graph, the gastric pH impact has lessened (Figure 12). At this 100-milligram dose and with gastric pH on the order of 4 to 5, there is still a large 75% absorbed projection for this molecule. When assessing fed-state physiology impact at a 400-milligram dose, there is a lessened impact of these different physiological changes (Figure 12, right graph). Presently, there is an amorphous, delayed-release tablet of posaconazole on the market. It doesn't release much of the drug in the stomach, which is contrary to our assumption for a simulation, but it does show that exposure with these amorphous tablets is increased threefold in the fasted state compared to NOXAFIL OS®. The food effect is significantly lessened, a 1.1- to 1.5-fold increase, and the exposure of ARAs is completely mitigated.
Based on these projections, a posaconazole ASD amorphous solid dispersion tablet was developed. We had a relatively high drug loading (75%) in an Eudragit® amorphous solid dispersion that was granulated with hydroxypropyl methylcellulose acetate succinate (HPMCAS-H) to maintain supersaturated drug concentrations and get a high drug loading on the tablet (25%). Looking at an immediate release tablet with rapid disintegration and the physical stability under accelerated conditions, we found the ASD to be physically stable. On the right side of Figure 13, the ASD tablet and NOXAFIL OS® suspension are compared in a gastric to intestinal transfer in vitro dissolution test under non-sink conditions. The ASD tablet outperformed NOXAFIL OS® by maintaining supersaturation at the amorphous solubility for the duration of the test.
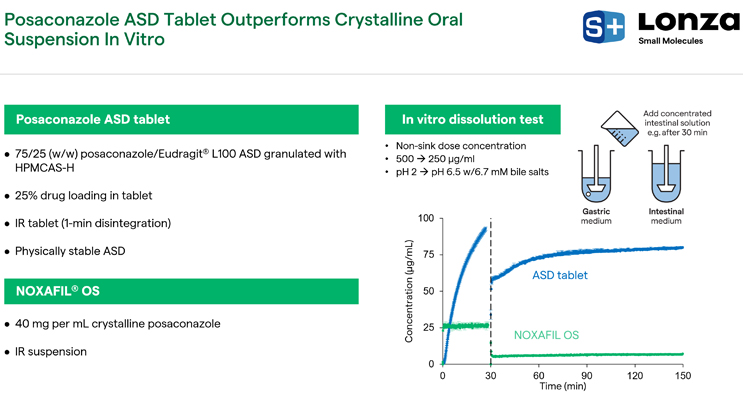
Figure 13. Posaconazole ASD tablet vs. NOXAFIL OS®
Next, PBPK modeling was used to de-risk an upcoming preclinical dog study by looking at two formulations, the ASD tablet and NOXAFIL OS®. In addition to the in vitro measured solubilities and API structure, we used observations from the dissolution test, Caco-2 permeation data, and in vivo IV bolus data from the literature to ground our permeation rate in dogs and understand our clearance and volume of distribution to predict plasma profiles. ADMET Predictor® and GastroPlus® were utilized to generate the following outputs: predicted plasma exposure, amorphous enhancement, and sensitivity to physiological and formulation variables.
The GastroPlus® model targeted fasted dog physiology at a 100-milligram dose, making bottom-up predictions with no optimization. We did not do any curve fitting; instead, we relied on mechanistic inputs derived from ADMET Predictor® or measured. In Figure 14 on the upper right plot, the amorphous form is not sensitive to precipitation time, whereas the crystalline NOXAFIL® is very sensitive, and there is an increase in exposure for the amorphous form (Figure 14). In the bottom plot of Figure 14, the amorphous form is projected to be resistant to stomach pH, whereas the crystalline NOXAFIL® suspension is still very sensitive, as reflected with human projections. This demonstrates that the amorphous form is projected to reduce sensitivity to these physiological and formulation variables. We project the AUC to be enhanced anywhere from two- to 15-fold compared to the crystalline suspension. The magnitude of that increase depends on the gastric pH and the precipitation time of NOXAFIL OS®.
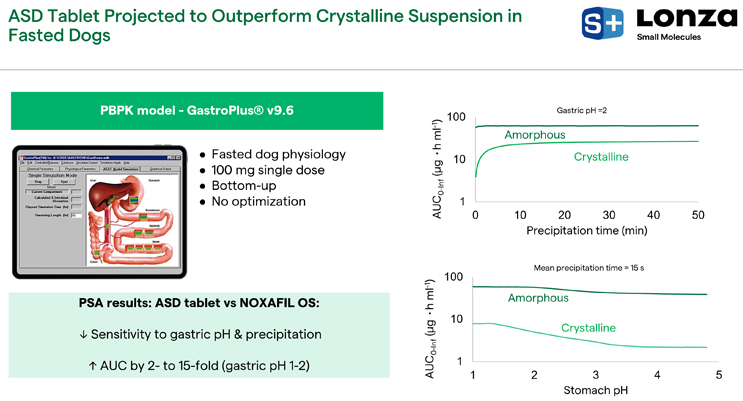
Figure 14. ASD vs. crystalline suspension in fasted dogs
After conducting an in vivo study in fasted beagle dogs who had been administered pentagastrin to reduce their stomach pH to 1 to 2 pH, the ASD tablet outperformed the crystalline suspension in terms of area of the curve by twofold (Figure 15). Our PSAs projected this, but since it was only a twofold enhancement, it is likely that the stomach pH of the beagle dogs was closer to 1 and precipitation time of posaconazole was relatively long, meaning that super saturation may have been achieved for about 1 hour. As such, the ASD tablet outperforms posaconazole in ideal state conditions, i.e., with a reduced stomach pH. We would expect a higher magnitude of exposure increase under higher gastric pH conditions (e.g., in the presence of an ARA).
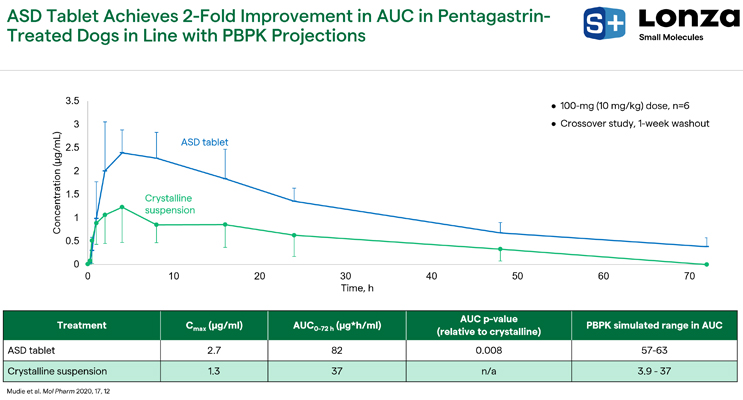
Figure 15. ASD tablet vs. crystalline suspension results
In summary, we successfully used PBPK modeling and in-house in vitro tools to identify poor oral absorption of posaconazole a priori. The amorphous form was forecasted as a viable strategy to increase absorption and decrease sensitivity to physiological variables with milligrams of API in a one-week duration without in vivo data. Once we had identified the amorphous form as a mitigation strategy, we developed a robust posaconazole amorphous solid dispersion tablet that outperformed the crystalline suspension in beagle dogs. PBPK was used to set expectations for the dog study by forecasting exposure enhancement of ASD compared to NOXAFIL OS® with grams of API in about three months, which mainly reflects the time it took to develop the spray dried amorphous dispersion and the tablet formulation. Preclinical study data helped us understand metabolism and elimination parameters of posaconazole.
Utilize PBPK to Improve Results and Save Money
PBPK studies can be conducted on rapid timelines with minimal amounts of API. The findings help drug sponsors make informed decisions about the priorities of their drug development process, gain improved context for understanding poor absorption, and implement risk mitigation strategies early on. Ultimately, PBPK modeling helps save time and money by reducing the need for reformulation or repeated in vivo studies. Lonza estimates PBPK savings of $500,000 to $2 million and a timeline reduction of six to nine months. With PBPK, you can improve productivity, increase your chances of being first to market, and help ensure vital medicines reach the patients who need them.